Translating Research Into Practice
Clinical Trial Design and Drug Approval in Oncology: A Primer for the Advanced Practitioner in Oncology
Sandra E. Kurtin,(1) PhD, ANP-C, AOCN®, and Rashida Taher,(2) MPH, PA-C
From (1)The University of Arizona Cancer Center, Tucson, Arizona; (2)Lifespan Cancer Institute, Providence, Rhode Island
Authors’ disclosures of conflicts of interest are found at the end of this article.
Correspondence to: Sandra Kurtin, PhD, ANP-C, AOCN®, The University of Arizona Cancer Center, 3838 N. Campbell Avenue, Tucson, AZ 85719-1454. E-mail: sandra.kurtin@bannerhealth.com
J Adv Pract Oncol 2020;11(7):736–751 |
https://doi.org/10.6004/jadpro.2020.11.7.7 |
© 2020 Harborside™

 ABSTRACT
ABSTRACT
Evidenced-based practice requires timely and accurate integration of scientific advances. This presents a challenge for the oncology clinician given the robust pace of scientific discovery and the increasing number of new drug approvals and expanded indications for previously approved drugs. All currently available antineoplastic therapies have been developed through the clinical trials process. Advanced practitioners (APs) in oncology are often involved in the conduct of clinical trials as primary investigators, sub-investigators, study coordinators, or in the delivery and monitoring of care to patients enrolled in these trials. A prerequisite to evidenced-based practice is understanding how clinical trials are conducted and how to critically analyze published results of studies leading to U.S. Food & Drug Administration approval. Any AP involved in the clinical management and supportive care of patients receiving antineoplastic therapies should be able to critically review published data to glean findings that warrant a change in practice. The goals of this manuscript are to summarize key elements of the clinical trial process for oncology drug development and approval in the United States and to provide a primer for the interpretation of clinical data.

ARTICLE
Translating research into clinical practice is a challenging and valuable skill for the advanced practitioner (AP) in oncology. Recent analysis of 585 trials registered in the National Cancer Institute Clinical Trials database showed that 29% remained unpublished within 5 years of completion of the study (Jones et al., 2013). A second study showed that among 1,075 abstracts describing 378 randomized and 697 nonrandomized clinical trials presented as abstracts at the American Society of Clinical Oncology (ASCO) meeting, only 75% of randomized and 54% of nonrandomized trials (61% overall) were ever published (Massey, Wang, Prasad, Bates, & Fojo, 2016). The average time from completion of a trial and publication among 809 trials closed to accrual between 2009 and 2013 was 47 months, with only 18.8% of these being published within 2 years of trial completion (Chapman et al., 2017). Unpublished results limit our ability to integrate the findings of these trials into practice, favorable or unfavorable, and diminish the efforts made by patients participating in those trials. Only 2% to 4% of all adults with cancer participate in clinical trials, the majority being under the age of 65 (Denson & Mahipal, 2014). Given the small number of patients participating in clinical trials today, publication of results should be a priority. Furthermore, the robust pace of scientific discovery in oncology places renewed emphasis on the ability of clinicians and scientists to critically review published data to allow appropriate and effective integration of the findings into the mainstream delivery of care.
Advanced practitioners in oncology are involved in the clinical management and supportive care of patients receiving antineoplastic therapies. Advanced practitioners are often involved in the conduct of clinical trials as primary investigators (PIs), sub-investigators, study coordinators, or in the delivery and monitoring of care to patients. Although the work is arduous, requiring meticulous assessment, documentation, and adherence to the protocol design, bringing a new therapy to trial and in some cases to market is one of the greatest privileges in oncology practice. Understanding evidence-based practice requires the systematic analysis of the scientific process evident in published works. The ability to independently, critically, and effectively articulate published data allows the AP the ability to glean findings that warrant a change in practice. Fortunately, summaries provided by various professional organizations, educational groups, or pharmaceutical companies allow for rapid dissemination of clinical trial results. However, the ability to independently review and effectively articulate trial results to colleagues and patients is an invaluable skill for APs. Concepts from the peer review and journal club processes offer practical and well-established criteria for critical appraisal of scientific literature.
The goals of this manuscript are to summarize key elements of the clinical trial process for new oncology drug development and approval in the United States and to provide a primer for the interpretation of clinical data. Part two and three of this series will apply this information to recent U.S. Food & Drug Administration (FDA) approvals in oncology with discussion of the implications for practice in both hematologic malignancies and solid tumors.
Key Elements of Clinical Trials Considered in Drug Approval
Clinical trials are used to test drugs, vaccines, and other medical interventions. Each clinical trial starts with a question or hypothesis generated based on clinical expertise, collaboration, and review of extant literature. It is incumbent on the PI(s) to select a question that will expand knowledge, offer clinical benefit to study participants (efficacy) and improve or sustain quality of life (safety). The trial design will be dictated by the question at hand and the available relevant science. The phase of clinical trial is indicative of the question at hand and the state of science relative to that question (Table 1). The phase of trial indicates the maturity of the science relative to the stated hypothesis and most often guides the study design (Table 2).
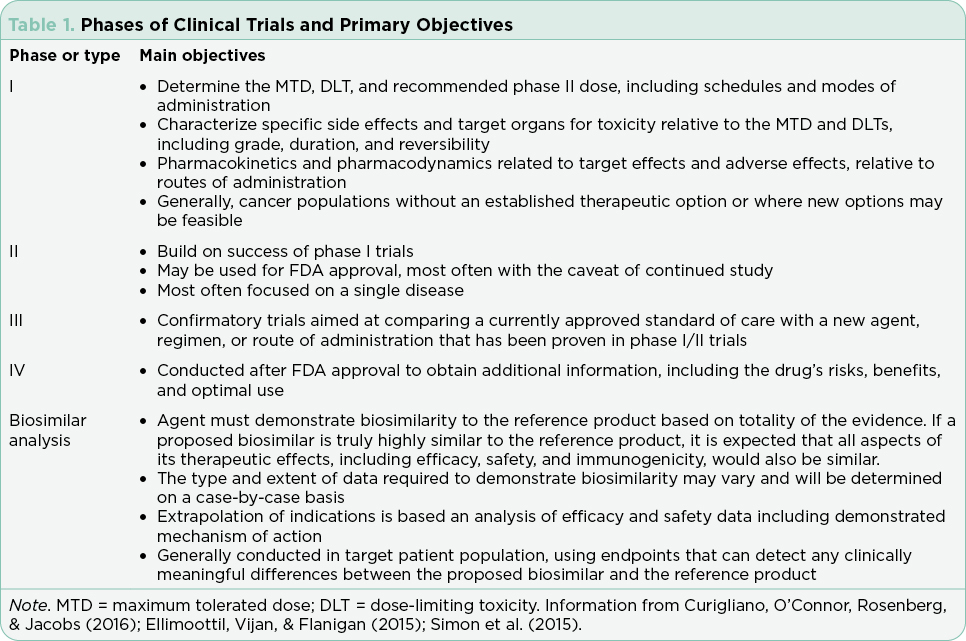
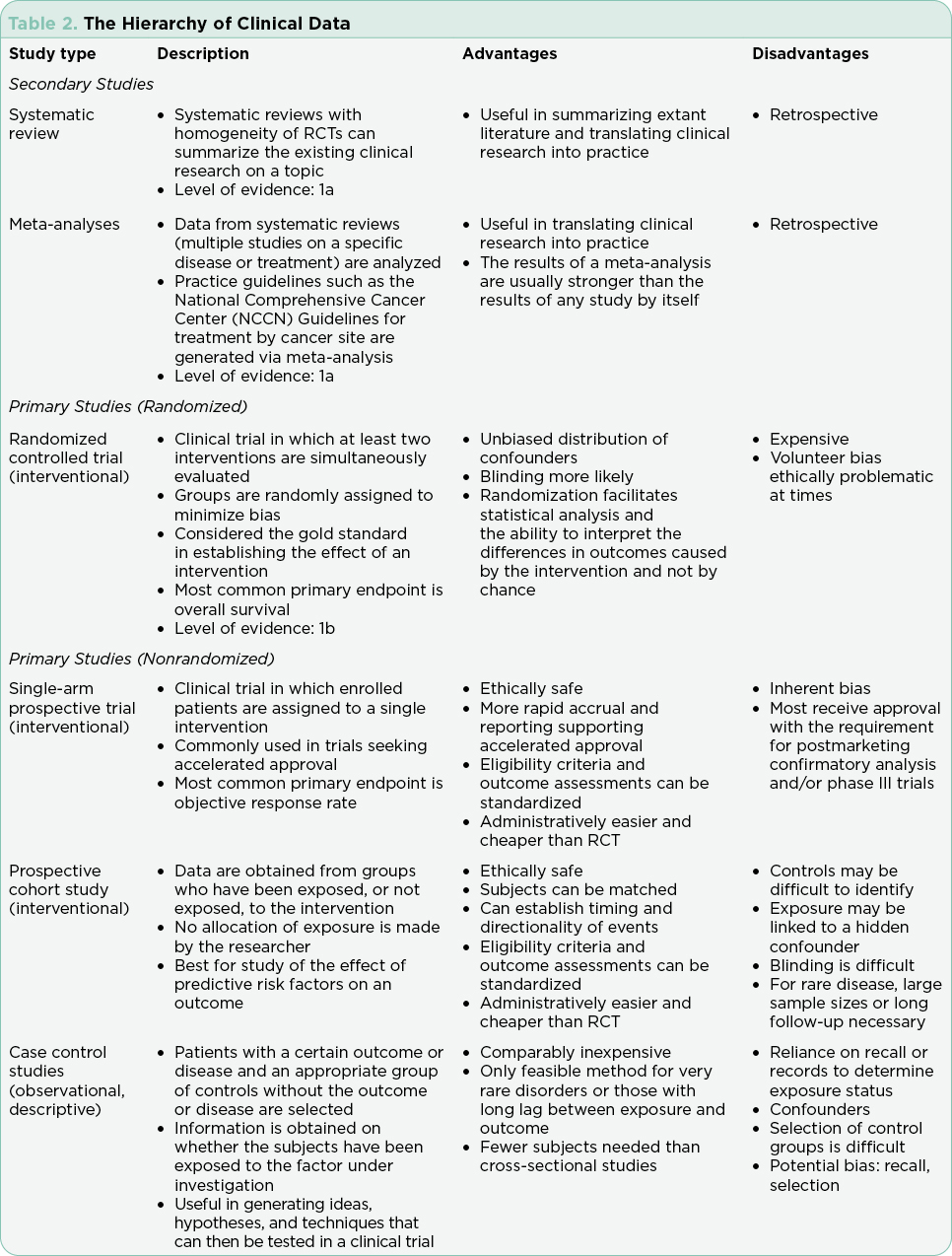

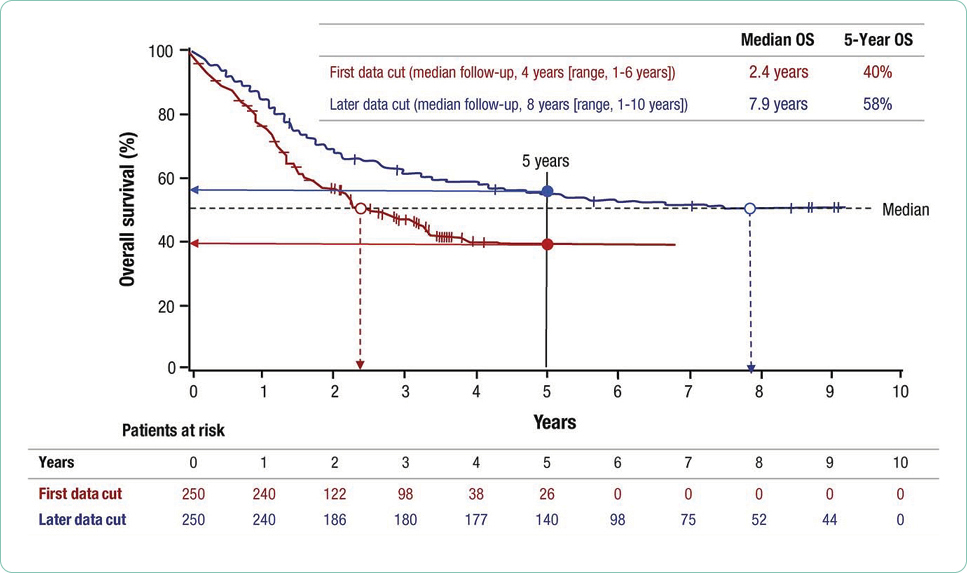
Interventional trials provide the foundation for new drug approvals in the United States. The randomized controlled trial (RCT) provides the most precise, thorough, and reliable characterization of therapeutic interventions by limiting bias through randomization (Simon et al., 2015). Blinded RCTs reduce bias further as clinicians are not told whether the patient will receive drug A or drug B. If there is a significant benefit in one arm of the trial at a predefined interim data analysis, crossover to the arm using the newer treatment may be allowed, and the trial will be unblinded at that time. Trials that include a crossover design with unblinding where clinicians may be more inclined to take patients off the control arm may confound analysis of progression-free survival (PFS; Simon et al., 2015). This is in part due to the challenges of recruitment but may also be driven by the selected endpoints and the time required to enroll enough subjects to reach statistical significance. Overall survival (OS) is generally the gold standard for the primary endpoint in RCTs, although reaching statistically significant OS can take many years in selected diseases. Demonstrating a survival advantage is logistically difficult in diseases with relatively favorable overall survival (patients lost to follow-up) and is very costly. As a result, PFS has been increasingly used as a surrogate to OS to allow earlier drug approval with many trials continuing their analysis post approval to achieve OS data. It is important to understand the differences and implications of different primary and secondary endpoints with regard to drug approval.
Although the RCT is considered the gold standard, there are instances in which this study design is not feasible. These include trials for drugs that target rare cancers where there are limited available therapies, trials in which there is a known actionable biomarker, and trials where the currently available therapies are highly toxic and/or marginally effective (Simon et al., 2015).
Single-arm trials, although limited by inherent bias, are often used in these cases. Single-arm trials generally focus on overall response rates based on the understanding that without treatment, the underlying disease would inevitably progress. Effective single-arm trials often move on to a randomized trial for validation. All RCTs and single-arm interventional trials are built on the foundation of early phase trials necessary for drug development. The rapid expansion of tailored therapies, with proof of therapeutic concept verified in laboratory models using cell lines derived from the science of cloning, has led to an increase in first-in-human trials approved by the FDA (Prowell, Theoret, & Pazdur, 2016). Rapid expansion of dosing cohorts to establish maximum tolerated dose and efficacy endpoints more efficiently are strategies used to accelerate the drug development process in these trials (Mayawala, Tse, Rubin, Jain, & de Alwis, 2017).
Most recently, noninferiority trials have emerged to substantiate alternative routes of administration, schedules for administration, and biosimilar agents. The standards for noninferiority trials are different than those for RCTs such that the design and analysis of outcomes must be evaluated within the context of each trial (Dunn, Copas, & Brocklehurst, 2018; Ganju & Rom, 2017). It is not within the scope of this paper to address the scientific principles and processes used in the development and approval of biosimilar agents. The reader is referred to several recent publications (Nabhan, Parsad, Mato, & Feinberg, 2018; Rifkin & Peck, 2017).
Inclusion and exclusion criteria must be defined based on established literature and clinical practice guidelines to ensure a homogenous study group and exclude patients who are at increased risk for adverse events based on what is known about the drug regimen and the disease being investigated. It is important to note that these patients, in most cases, are not representative of the general population where the presence of comorbidities or other factors excluded in the trial are prevalent. More recently, therapies tailored to specific targets or pathways have limited inclusion criteria such that recruitment may take a prolonged period of time. It is critical for clinicians to consider the inclusion and exclusion criteria in the postmarketing setting to safely integrate new therapies into practice with patients who may be at a higher risk for adverse events.
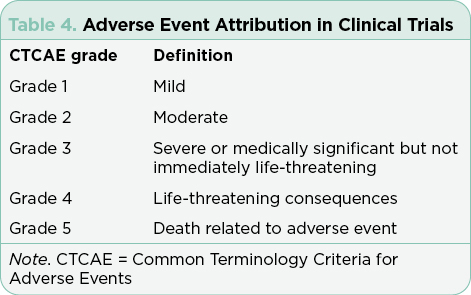
Safety must be evaluated in every study prior to FDA approval. The Common Terminology Criteria for Adverse Events (CTCAE) are applied across studies to summarize tolerability, safety, and patient-reported outcomes systematically (Shepshelovich et al., 2019; U.S. Department of Health and Human Services, 2017). The CTCAE criteria are subdivided by organ system and common adverse events. Each adverse event (AE) is graded on a scale of 1 (minor) to 5 (death). This process is critical in interpreting the risks and benefits of each treatment and is used to develop the AE profiles printed in package inserts for drugs approved by the FDA. The CTCAE has published multiple versions, and it is important to note the version being used in the trial to effectively interpret the AEs reported. To reduce the subjectivity inherent in clinicians’ attribution of AEs, patient-reported outcomes (PRO) have gained importance to better describe safety and tolerability across the life of a trial (Kluetz, Chingos, Basch, & Mitchell, 2016). The Patient-Reported Outcomes Version of the CTCAE (PRO-CTCAE) incorporates 78 patient-reported symptomatic AEs to collect subjective symptoms directly from patients, including measures for health-related quality of life (HRQOL).
Clinical Trial Endpoints
Researchers must select primary and secondary endpoints for each study. The primary endpoint is the major focus of the study and is essentially what is expected to happen or the primary aim of the study. Secondary endpoints are outcomes expected to add to the significance of the new therapy being investigated. These endpoints are selected based on analysis of the existing literature, treatment landscape, the anticipated fit of the treatment being studied, power analysis, and the research interests and expertise of the research team. Overall survival, PFS, PFS2, event-free survival (EFS), objective response rate, duration of response (DOR), time to progression (TTP), time to treatment failure (TTF), and depth of response are the most common primary and secondary endpoints (Table 3; Hickey et al., 2013). Patient-centered endpoints include OS and HRQOL, both affecting a patient’s feeling of well-being and directing patient clinical benefit. Tumor or disease-specific endpoints include PFS or depth of response. Although OS is considered the hallmark of success, data to support statistically significant improvement of OS may require years of data collection/monitoring based on the expected survival in selected tumor types; PFS always precedes OS and has become a more common primary endpoint in clinical trials today. If PFS or TTP is selected as a primary endpoint, OS should be reported as a secondary endpoint and vice versa. Importantly, OS reporting may be confounded by subsequent therapies (Estey, Othus, Lee, Appelbaum, & Gale, 2016). In the era of accelerated approvals, overall response rate is the standard for a primary endpoint (Blumenthal et al., 2015; Chen, Raghunathan, & Prasad, 2019).

Criteria for response should be based on various disease-specific working group consensus statements. These must be articulated prior to study approval. These criteria provide the foundation for claims of efficacy. The specific response criteria and version must be noted, as these criteria are regularly updated based on emerging data. The ability to generalize results will be limited by the study design. Comparing two published trials with similar but not exactly the same methods and sample is not recommended.
The shift toward precision medicine, immunotherapies, and other treatments with novel mechanisms of action has shifted the traditional definition of progression and response. The phenomenon of pseudoprogression seen in trials using immunotherapies has required redefinition of progression (Hochmair, Schwab, Burghuber, Krenbek, & Prosch, 2017). For patients with rare or extensive disease, stable disease or even a slow progression is considered acceptable outside of a clinical trial (Kaufmann, Pariser, & Austin, 2018). Similarly, the integration of depth of response, specifically achieving undetectable minimal residual disease status in hematologic malignancies has been correlated with PFS and OS and is now being integrated into the response criteria for selected diseases (Gormley et al., 2016; Hallek et al., 2018; Heuser, Mina, Stein, & Altman, 2019; Kovacs et al., 2016; Medeiros, 2018; Molica, Giannarelli, & Montserrat, 2019). Although these new criteria have been defined, the application in general practice for patients treated outside of a clinical trial is not clearly defined in many cases. Caution is recommended prior to changing therapies where there is no clear data to support loss of undetectable minimal residual disease as progression.
The duration of response, although not often a primary endpoint, is critical to evaluating treatment options over time. The durability of a treatment option including tolerability is often lost in a new drug approval where the data cutoff points are focused on achievement of the primary and secondary endpoints to support approval. Both long-term responses, survival, and late relapses are not captured in trials that do not monitor patients over time or where patients are lost to follow-up (Cuzick, 2015). In addition, many blinded trials are unblinded at predetermined data cutoff points to facilitate statistical analysis, making long-term analysis of outcomes more difficult. Case reports and anecdotal data are often the mainstay for reporting outcomes in the subset of patents that achieve the most durable responses to investigational agents that are subsequently approved (Kurtin & List, 2009).
Conduct of Clinical Trials
A site initiation visit is conducted by the study monitor to ensure that all clinicians and clinical trials staff are registered on the study, fully trained on all aspects of the study, and understand the processes for conducting the study, including reporting of unexpected adverse events or deviations. Consent forms, data monitoring processes and forms, and safety requirements are outlined in detail. Once the study is open at an individual site, patient accrual begins. Patient recruitment requires a coordinated team approach to inform clinicians about the trial and facilitate patient screening. This requires a level of knowledge on the part of the primary clinical team to effectively articulate the value of a clinical trial in the treatment selection process to the patient and their caregivers. If a patient is eligible for a trial, it is always a preferred option, as accessibility and eligibility for trials can change quickly. Applying the mantra “Never exclude a treatment option” will offer the patient the best opportunity to receive all available therapies.
For those patients interested in participating in a clinical trial, a detailed process of screening ensures application of the predefined inclusion and exclusion criteria. If eligible, the clinical trial is then submitted for insurance approval for the individual patient. Unfortunately, despite federal legislation, not all insurance plans approve investigational treatment. The number of open trials in the United States and the number of FDA approvals in oncology over the past decade contribute to challenges in the recruitment of patients. In many instances, patients are not referred to tertiary centers for clinical trial participation if FDA-approved drugs are available for use.
Each trial includes a schema, or a calendar of events with a detailed step-by-step process for conducting the trial. The schema includes any diagnostic imaging, laboratory or tissue testing, clinical visits, and medication reconciliation. Also included in most studies are patient journals to facilitate self-reporting of any adverse events. Physical exams and evaluation of AEs are included in these prespecified visits and must also be reported for any unexpected visits or admission to urgent care, the emergency room, or to the inpatient setting. Patients are queried for HRQOL indices at prespecified intervals to evaluate patient-reported outcomes. Reviewing the schema and the intensity of visits is critical to guide the patient in making the decision to participate. For some patients, the intensity of a trial, particularly phase I trials that require frequent pharmacokinetic testing, may dissuade the patient from participation.
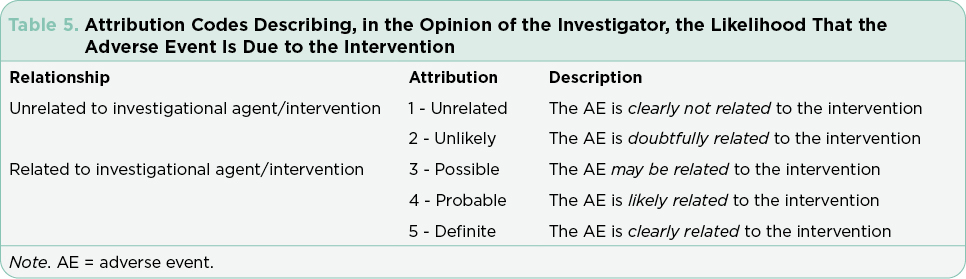
Safety monitoring is at the core of managing patients on clinical trials and requires attention to detail. Advanced practitioners are often designated as Co-PIs or sub-investigators on these trials and see patients for trial-related visits. These visits require a detailed review of systems to capture any changes from baseline, details of any new clinical or symptom findings, and capture of any missed doses or medications added for symptom management. Adverse events must be attributed as to system, causality, and estimated severity (Tables 4 and 5). These attributions are the primary source for reporting safety and are used to create the tables and charts included in package inserts for FDA-approved drugs. Patients who require urgent or emergent care or hospitalization require immediate reporting and are automatically considered to have serious adverse events that must be reported to the medical monitor for the study within 24 to 48 hours. This requires a coordinated effort on the part of the research team, the patient, and their caregivers. Accurate and timely reporting is essential to avoid adverse events related to the investigational regimen across all study sites. Despite ongoing efforts to refine the definitions to limit variability in attributions, the system remains imperfect and open to subjective interpretation (George et al., 2019). Postmarketing reporting is critical to integrate continued observations for approved drugs, flag any sentinel events, and continue to ensure the safety of the public at large.
The FDA Approval Process
Prior to 1992, FDA approval was only granted through the standard process for review and in the majority of cases required completion of a RCT. The FDA established the Oncology Center of Excellence (OCE) in January 2017 to streamline the development of cancer therapies through expedited review of drugs, biologics, and devices (Goldberg, Blumenthal, McKee, & Pazdur, 2018). The accelerated approval pathway utilizes surrogate measures such as biomarkers, objective overall response, and in some cases, clinical benefit (Chen et al., 2019; Gyawali, Hey, & Kesselheim, 2019). This is in response to the transformative effect of biomarker-driven therapies, the field of immunology, and the desire to make these drugs available to the public.
Between December 11, 1992, and May 31, 2017, the FDA granted accelerated approval for drugs considered to be transformative in areas of high unmet need based on phase II data and on endpoints other than OS (Beaver et al., 2018; Bloomfield et al., 2018). Among 64 malignant hematology and oncology products for 93 new indications, the most common primary endpoints included response rate (n = 81, 87%), PFS or TTP (n = 8, 9%), and disease-free survival (n = 4, 4%). Importantly, single-arm trial designs were the most common (n = 67, 72%; Beaver et al., 2018). For many trials with accelerated approval, postmarketing validation via long-term follow-up or conduct of a phase III randomized trial may be required for continued approval. In this analysis, at a median of 3.4 years after the accelerated approval, 55% (n = 51) had fulfilled their postmarketing requirement and verified benefit, 40% (n = 37) had not yet completed the confirmatory trials, and 5% (n = 5) had been withdrawn from the market.
Interpretation of Study Results: Efficacy
The efficacy of each trial is based on meeting both primary and secondary endpoints defined prior to submission for Institutional Review Board approval. Although the science sometimes moves faster than the enrollment process as previously discussed, the primary hypotheses of the study cannot be modified. The most common strategies for communicating outcomes include both statistical descriptions and graphic display (Table 6). Understanding how to interpret written and graphical displays of data is essential to critical review and application of the study results to practice. Specific examples of recent FDA-approved therapeutics for hematologic malignancies and the published pivotal trials will be presented in part two of this series. Specific examples of recent FDA-approved therapeutics for solid tumors and the published pivotal trials will be presented in part three of this series.

Interpretation of Study Results: Safety
Efficacy without safety is not an acceptable outcome in clinical trials or in standard of care treatment. The CTCAE reporting structure (Table 4) in clinical trials has required continued editing to reflect knowledge gained across medical specialties relevant to organ function, degree of organ damage, and the potential causative agent. Publication of pivotal trials provides the most complete publicly available resource for summarizing adverse events. Reading the package insert and the published registration trial is recommended to integrate new therapies into practice and to guide estimates of risk and benefits. Understanding the grading system for AEs, the criteria for specific toxicities, and the population studied in the pivotal trial will improve APs’ ability to safely integrate new therapies into practice. Importantly, package inserts include all trials registered for the approved agent and must be interpreted in the context of the specific diseases and any combination regimens.

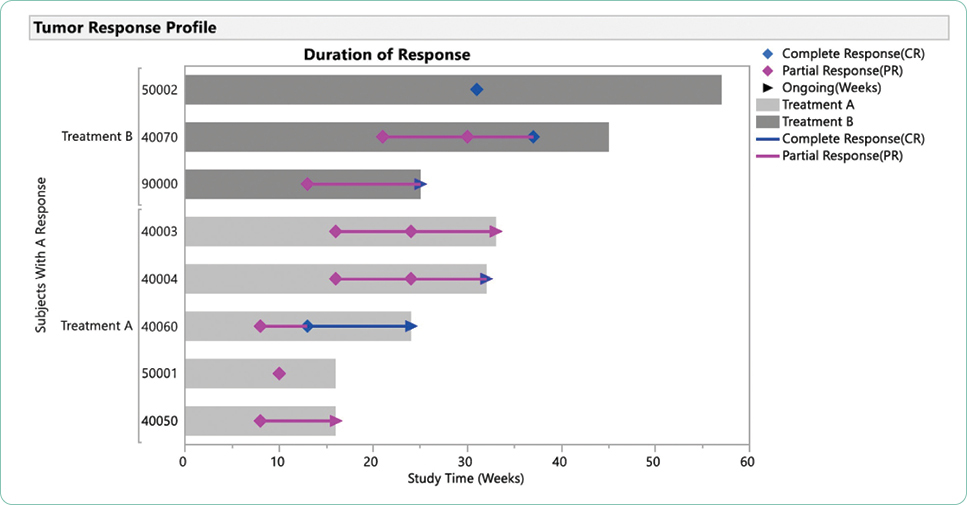
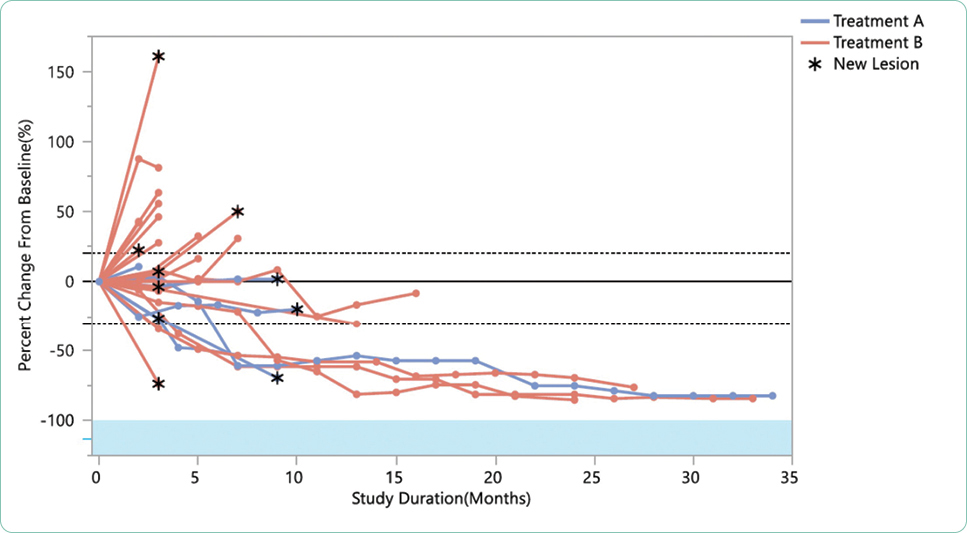
Adverse events are generally summarized in publications and in the package insert as all-grade or grade ≥ 3. Grade 1 AEs are generally bothersome but not life-threatening and may or may not require interventions but should be monitored closely to avoid more severe AEs. Grade 2 AEs may require interventions to control symptoms, but do not generally require dose modifications. Grade 3 or 4 AEs represent serious toxicities that require careful assessment of cause and consideration of options for mitigation and management. In some cases, the drug will need to be held, dose reduced, or discontinued based on the individual guidelines for that drug and regimen. Unfortunately, many treatments are prematurely discontinued without application of all available strategies available for mitigation and management, effectively limiting potential benefit due to the fear of AEs.
Given the inherent flaws of clinician-only reporting of AEs using the CTCAE criteria, the PRO-CTCAE has been developed (Kluetz et al., 2016). Familiarity with the PRO-CTCAE will become a necessary component of conducting clinical trials in oncology to optimize benefit and limit risk. Adverse events attributed to immunotherapies have required development of specific criteria to estimate severity (Michot et al., 2016; Wang & Xu, 2019; Yu et al., 2019).
Implications for the AP
All currently available therapies for cancer patients have been derived through the conduct of clinical trials. Advanced practitioners in oncology play an integral role in the conduct of clinical trials and in the integration of new therapies into mainstream practice. Review and synthesis of the plethora of clinical trial outcome data has become an arduous yet essential task. Applying general concepts and strategies for review will assist in the interpretation and clinical application of the emerging data. Familiarity with the hierarchy of clinical trial design, the shifting of desired endpoints in the era of precision medicine and robust clinical development, and the necessity of incorporating patient-reported outcomes will be necessary for all oncology clinicians, including the AP. For those APs involved in the conduct of clinical trials, familiarity with new definitions of response, changes in the attestation of AEs, including patient-reported outcomes, will be imperative to the effective conduct and reporting of the trial. There is an opportunity for all APs in oncology to play a larger role in the reporting of adverse events and development of strategies for the management of AEs in the postmarketing phase.
Disclosure
The authors have no conflicts of interest to disclose.
References
Beaver, J. A., Howie, L. J., Pelosof, L., Kim, T., Liu, J., Goldberg, K. B.,...Kluetz, P. G. (2018). A 25-year experience of US Food and Drug administration accelerated approval of malignant hematology and oncology drugs and biologics: A Review. JAMA Oncology, 4(6), 849–856. https://doi.org/10.1001/jamaoncol.2017.5618
Bloomfield, C. D., Estey, E., Pleyer, L., Schuh, A. C., Stein, E. M., Tallman, M. S., & Wei, A. (2018). Time to repeal and replace response criteria for acute myeloid leukemia? Blood Reviews, 32(5), 416–425. https://doi.org/10.1016/j.blre.2018.03.006
Blumenthal, G. M., Karuri, S. W., Zhang, H., Zhang, L., Khozin, S., Kazandjian, D.,...Pazdur, R. (2015). Overall response rate, progression-free survival, and overall survival with targeted and standard therapies in advanced non-small-cell lung cancer: US Food and Drug Administration trial-level and patient-level analyses. Journal of Clinical Oncology, 33(9), 1008–1014. https://doi.org/10.1200/jco.2014.59.0489
Chapman, P. B., Liu, N. J., Zhou, Q., Iasonos, A., Hanley, S., Bosl, G. J., & Spriggs, D. R. (2017). Time to publication of oncology trials and why some trials are never published. PLoS One, 12(9), e0184025. https://doi.org/10.1371/journal.pone.0184025
Chen, E. Y., Raghunathan, V., & Prasad, V. (2019). An overview of cancer drugs approved by the US Food and Drug Administration based on the surrogate end point of response rate. JAMA Internal Medicine, 179(7), 915–921. https://doi.org/10.1001/jamainternmed.2019.0583
Curigliano, G., O’Connor, D. P., Rosenberg, J. A., & Jacobs, I. (2016). Biosimilars: Extrapolation for oncology. Critical Reviews in Oncology/Hematology, 104, 131–137. https://doi.org/10.1016/j.critrevonc.2016.06.002
Cuzick, J. (2015). Statistical controversies in clinical research: Long-term follow-up of clinical trials in cancer. Annals of Oncology, 26(12), 2363–2366. https://doi.org/10.1093/annonc/mdv392
Denson, A. C., & Mahipal, A. (2014). Participation of the elderly population in clinical trials: Barriers and solutions. Cancer Control, 21(3), 209–214. https://doi.org/10.1177/107327481402100305
Dunn, D. T., Copas, A. J., & Brocklehurst, P. (2018). Superiority and non-inferiority: Two sides of the same coin? Trials, 19(1), 499. https://doi.org/10.1186/s13063-018-2885-z
Ellimoottil, C., Vijan, S., & Flanigan, R. C. (2015). A primer on clinical trial design. Urologic Oncology, 33(3), 116–121. https://doi.org/10.1016/j.urolonc.2014.12.014
Estey, E., Othus, M., Lee, S. J., Appelbaum, F. R., & Gale, R. P. (2016). New drug approvals in acute myeloid leukemia: What’s the best end point? Leukemia, 30(3), 521–525. https://doi.org/10.1038/leu.2015.262
Fiteni, F., Westeel, V., Pivot, X., Borg, C., Vernerey, D., & Bonnetain, F. (2014). Endpoints in cancer clinical trials. Journal of Visceral Surgery, 151(1), 17–22. https://doi.org/10.1016/j.jviscsurg.2013.10.001
Ganju, J., & Rom, D. (2017). Non-inferiority versus superiority drug claims: the (not so) subtle distinction. Trials, 18(1), 278. https://doi.org/10.1186/s13063-017-2024-2
George, G. C., Barata, P. C., Campbell, A., Chen, A., Cortes, J. E., Hyman, D. M.,...Hong, D. S. (2019). Improving attribution of adverse events in oncology clinical trials. Controversy, 76, 33–40. https://doi.org/10.1016/j.ctrv.2019.04.004
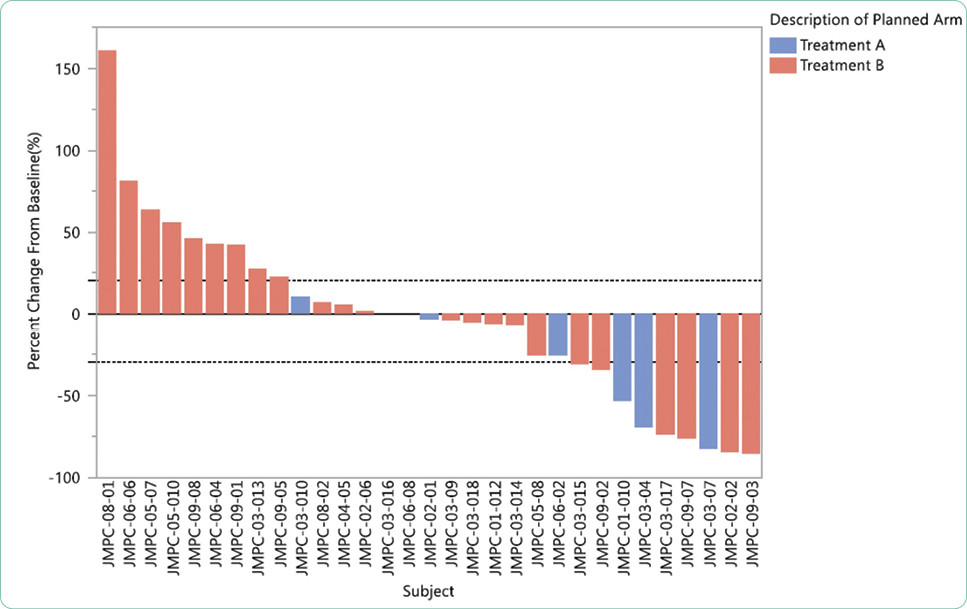
Gillespie, T. (2012). Understanding waterfall plots. Journal of the Advanced Practitioner in Oncology, 3(2), 106–111. https://doi.org/10.6004/jadpro.2012.3.2.6
Goldberg, K. B., Blumenthal, G. M., McKee, A. E., & Pazdur, R. (2018). The FDA Oncology Center of Excellence and precision medicine. Experimental Biology and Medicine (Maywood), 243(3), 308–312. https://doi.org/10.1177/1535370217740861
Gormley, N. J., Turley, D. M., Dickey, J. S., Farrell, A. T., Reaman, G. H., Stafford, E.,...Marti, G. E. (2016). Regulatory perspective on minimal residual disease flow cytometry testing in multiple myeloma. Cytometry Part B: Clinical Cytometry, 90(1), 73–80. https://doi.org/10.1002/cyto.b.21268
Gyawali, B., Hey, S. P., & Kesselheim, A. S. (2019). Assessment of the clinical benefit of cancer drugs receiving accelerated approval. JAMA Internal Medicine, 179(7), 906–913. https://doi.org/10.1001/jamainternmed.2019.0462
Hallek, M., Cheson, B. D., Catovsky, D., Caligaris-Cappio, F., Dighiero, G., Dohner, H.,...Kipps, T. J. (2018). iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood, 131(25), 2745–2760. https://doi.org/10.1182/blood-2017-09-806398
Heuser, M., Mina, A., Stein, E. M., & Altman, J. K. (2019). How precision medicine is changing acute myeloid leukemia therapy. American Society of Clinical Oncology Educational Book, 39, 411–420. https://doi.org/10.1200/edbk_238687
Hickey, R., Vouche, M., Sze, D. Y., Hohlastos, E., Collins, J., Schirmang, T.,...Salem, R. (2013). Cancer concepts and principles: primer for the interventional oncologist-part I. Journal of Vascular Interventional Radiology, 24(8), 1157–1164. https://doi.org/10.1016/j.jvir.2013.04.024
Ho, P. M., Peterson, P. N., & Masoudi, F. A. (2008). Evaluating the evidence: Is there a rigid hierarchy? Circulation, 118(16), 1675–1684. https://doi.org/10.1161/circulationaha.107.721357
Hochmair, M. J., Schwab, S., Burghuber, O. C., Krenbek, D., & Prosch, H. (2017). Symptomatic pseudo-progression followed by significant treatment response in two lung cancer patients treated with immunotherapy. Lung Cancer, 113, 4–6. https://doi.org/10.1016/j.lungcan.2017.08.020
Howick, J., Chalmers, I., Glasziou, P., Greenhalgh, J., Heneghan, C., Liberati, A.,…Thornton, H. (2011). The 2011 Oxford CEBM Evidence Levels of Evidence (Introductory Document). Retrieved from https://www.cebm.net/index.aspx?o=5653
Jones, C. W., Handler, L., Crowell, K. E., Keil, L. G., Weaver, M. A., & Platts-Mills, T. F. (2013). Non-publication of large randomized clinical trials: Cross sectional analysis. BMJ, 347, f6104. https://doi.org/10.1136/bmj.f6104
Kaufmann, P., Pariser, A. R., & Austin, C. (2018). From scientific discovery to treatments for rare diseases - the view from the National Center for Advancing Translational Sciences - Office of Rare Diseases Research. Orphanet Journal of Rare Diseases, 13(1), 196–196. https://doi.org/10.1186/s13023-018-0936-x
Kluetz, P. G., Chingos, D. T., Basch, E. M., & Mitchell, S. A. (2016). Patient-reported outcomes in cancer clinical trials: Measuring symptomatic adverse events with the National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). American Society of Clinical Oncology Educational Book, 35, 67–73. https://doi.org/10.14694/edbk_159514
Kovacs, G., Robrecht, S., Fink, A. M., Bahlo, J., Cramer, P., von Tresckow, J.,...Bottcher, S. (2016). Minimal residual disease assessment improves prediction of outcome in patients with chronic lymphocytic leukemia (CLL) who achieve partial response: Comprehensive analysis of two phase III studies of the German CLL Study Group. Journal of Clinical Oncology, 34(31), 3758–3765. https://doi.org/10.1200/jco.2016.67.1305
Kurtin, S. E., & List, A. F. (2009). Durable long-term responses in patients with myelodysplastic syndromes treated with lenalidomide. Clinical Lymphoma and Myeloma, 9(3), E10–E13. https://doi.org/10.3816/CLM.2009.n.053
Massey, P. R., Wang, R., Prasad, V., Bates, S. E., & Fojo, T. (2016). Assessing the eventual publication of clinical trial abstracts submitted to a large annual oncology meeting. Oncologist, 21(3), 261–268. https://doi.org/10.1634/theoncologist.2015-0516
Mayawala, K., Tse, A., Rubin, E. H., Jain, L., & de Alwis, D. P. (2017). Dose finding versus speed in seamless immuno-oncology drug development. Journal of Clinical Pharmacology, 57(S10), S143–S145. https://doi.org/10.1002/jcph.1001
Maymone, M. B. C., Gan, S. D., & Bigby, M. (2014). Evaluating the strength of clinical recommendations in the medical literature: GRADE, SORT, and AGREE. Journal of Investigative Dermatology, 134(10), 1–5. https://doi.org/10.1038/jid.2014.335
McKee, A. E., Farrell, A. T., Pazdur, R., & Woodcock, J. (2010). The role of the U.S. Food and Drug Administration review process: Clinical trial endpoints in oncology. Oncologist, 15(Suppl 1), 13–18. https://doi.org/10.1634/theoncologist.2010-S1-13
Medeiros, B. C. (2018). Interpretation of clinical endpoints in trials of acute myeloid leukemia. Leukemia Research, 68, 32–39. https://doi.org/10.1016/j.leukres.2018.02.002
Michot, J. M., Bigenwald, C., Champiat, S., Collins, M., Carbonnel, F., Postel-Vinay, S.,...Lambotte, O. (2016). Immune-related adverse events with immune checkpoint blockade: A comprehensive review. European Journal of Cancer, 54, 139–148. https://doi.org/10.1016/j.ejca.2015.11.016
Miclaus, K., & Li, L. (2018). Leveraging Standards for Effective Visualization of Early Efficacy in Clinical Trial Oncology Studies. Paper presented at the PharmaSUG 2018 China.
Molica, S., Giannarelli, D., & Montserrat, E. (2019). Minimal residual disease and survival outcomes in patients with chronic lymphocytic leukemia: A systematic review and meta-analysis. Clinical Lymphoma, Myeloma, and Leukemia, 19(7), 423–430. https://doi.org/10.1016/j.clml.2019.03.014
Nabhan, C., Parsad, S., Mato, A. R., & Feinberg, B. A. (2018). Biosimilars in oncology in the United States: A Review. JAMA Oncology, 4(2), 241–247. https://doi.org/10.1001/jamaoncol.2017.2004
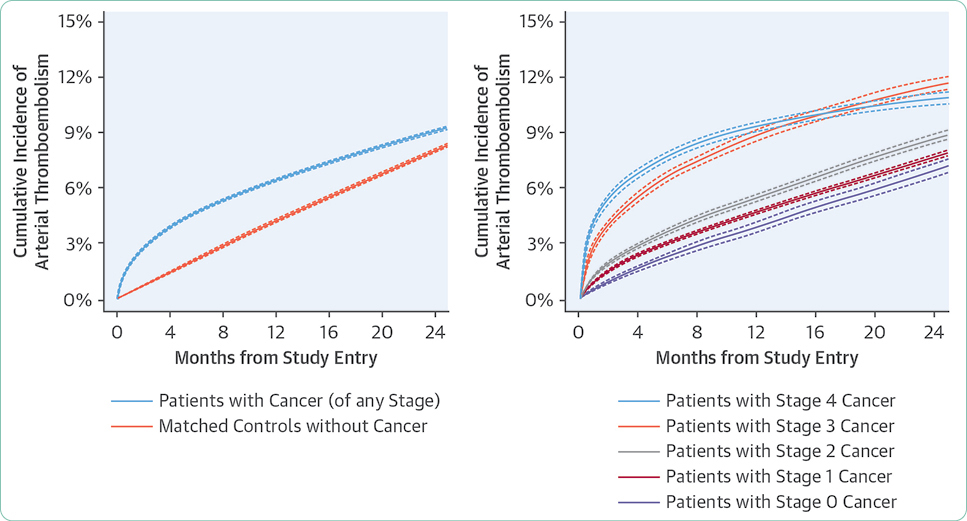
Navi, B. B., Reiner, A. S., Kamel, H., Iadecola, C., Okin, P. M., Elkind, M. S.,…DeAngelis, L. M. (2017). Risk of arterial thromboembolism in patients with cancer. Journal of the American College of Cardiology, 70(8), 926–938. https://doi.org/10.1016/j.jacc.2017.06.047.
Pocock, S. J., Clayton, T. C., & Stone, G. W. (2015a). Design of major randomized trials: Part 3 of a 4-part series on statistics for clinical trials. Journal of the American College of Cardiology, 66(24), 2757–2766. https://doi.org/10.1016/j.jacc.2015.10.036
Pocock, S. J., McMurray, J. J., & Collier, T. J. (2015b). Making sense of statistics in clinical trial reports: Part 1 of a 4-part series on statistics for clinical trials. Journal of the American College of Cardiology, 66(22), 2536–2549. https://doi.org/10.1016/j.jacc.2015.10.014
Pocock, S. J., McMurray, J. J. V., & Collier, T. J. (2015c). Statistical controversies in reporting of clinical trials: Part 2 of a 4-part series on statistics for clinical trials. Journal of the American College of Cardiology, 66(23), 2648–2662. https://doi.org/10.1016/j.jacc.2015.10.023
Prowell, T. M., Theoret, M. R., & Pazdur, R. (2016). Seamless oncology-drug development. New England Journal of Medicine, 374(21), 2001–2003. https://doi.org/10.1056/NEJMp1603747
Rifkin, R. M., & Peck, S. R. (2017). Biosimilars: Implications for clinical practice. Journal of Oncology Practice, 13(9_suppl), 24s–31s. https://doi.org/10.1200/jop.2017.025734
Sashegyi, A., & Ferry, D. (2017). On the interpretation of the hazard ratio and communication of survival benefit. Oncologist, 22(4), 484–486. https://doi.org/10.1634/theoncologist.2016-0198
Shepshelovich, D., McDonald, K., Spreafico, A., Razak, A. R. A., Bedard, P. L., Siu, L. L.,...Hansen, A. R. (2019). Feasibility assessment of using the complete Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) item library. Oncologist, 24(4), e146–e148. https://doi.org/10.1634/theoncologist.2018-0332
Simon, R., Blumenthal, G. M., Rothenberg, M. L., Sommer, J., Roberts, S. A., Armstrong, D. K.,...Pazdur, R. (2015). The role of nonrandomized trials in the evaluation of oncology drugs. Clinical Pharmacology & Therapeutics, 97(5), 502–507. https://doi.org/10.1002/cpt.86
U.S. Department of Health and Human Services. (2017). Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. Retrieved from https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm
U.S. Department of Health and Human Services. (2018). Clinical trial endpoints for the approval of cancer drugs and biologics: Guidance for industry. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics
van Rijn, M. H. C., Bech, A., Bouyer, J., & van den Brand, J. (2017). Statistical significance versus clinical relevance. Nephrology Dialysis Transplantation, 32(suppl_2), ii6–ii12. https://doi.org/10.1093/ndt/gfw385
Wang, Q., & Xu, R. (2019). Immunotherapy-related adverse events (irAEs): Extraction from FDA drug labels and comparative analysis. JAMIA Open, 2(1), 173–178. https://doi.org/10.1093/jamiaopen/ooy045
Yu, Y., Ruddy, K. J., Tsuji, S., Hong, N., Liu, H., Shah, N., & Jiang, G. (2019). Coverage evaluation of CTCAE for capturing the immune-related adverse events leveraging text mining technologies. AMIA Joint Summits on Translational Science, 2019, 771–778. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6568118/pdf/3056004.pdf